
Cannabigerol Benefits and Research
July 10, 2025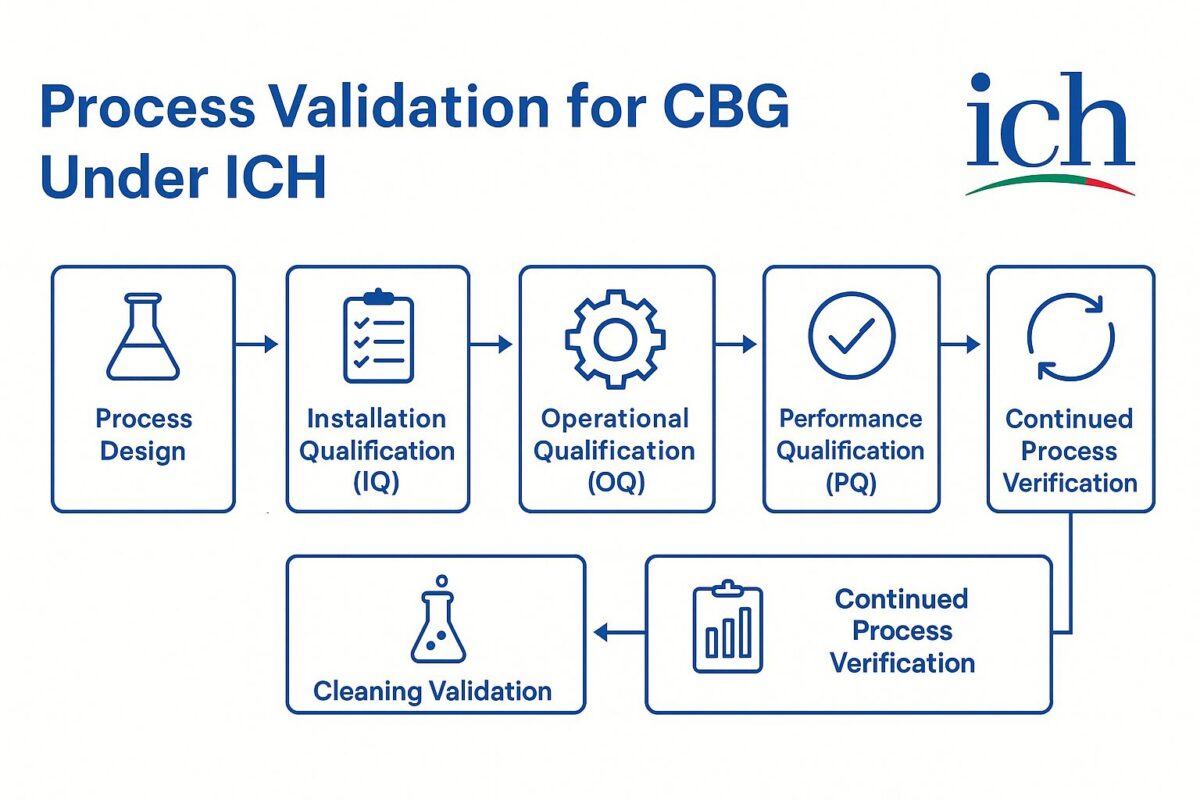
Process Validation and Stability Testing of Cannabigerol (CBG) Isolate under ICH Q7
Validating a novel cannabinoid such as cannabigerol (CBG) isolate as an Active Pharmaceutical Ingredient (API) under the ICH Q7 guidelines requires a rigorous and systematic approach. ICH Q7 provides a global cGMP framework for API manufacturing to ensure that APIs consistently meet defined quality and purity attributes. Drawing on experience from cannabidiol (CBD) – a more widely studied cannabinoid – many Good Manufacturing Practice principles are similar. However, CBG presents unique challenges because historically it has been a minor component of cannabis (often ~1% or less of the cannabinoid content) and thus lacks extensive prior data. This means the process validation and stability testing programs for CBG must be especially thorough to establish the product’s safety, quality, and shelf-life in the absence of extensive historical knowledge.
In the following sections, we outline the key steps in process validation for CBG isolate and the types of stability testing required, all in alignment with ICH Q7 and related ICH quality guidelines. We also discuss how lessons learned from CBD API production inform the CBG approach, and where CBG may require different considerations.
Process Validation of CBG Isolate under cGMP
Process validation is the documented evidence that a manufacturing process, when operated within specified parameters, can perform effectively and reproducibly to yield an API meeting its predetermined quality attributes. In the context of CBG isolate, a full lifecycle approach to process validation is followed, from initial process design through equipment qualification, performance qualification, and ongoing monitoring. By adhering to ICH Q7 and cGMP principles at each stage, manufacturers gain confidence that the process consistently produces CBG of suitable quality for pharmaceutical use. Below are the key steps in the process validation sequence:
-
Process Design: This initial stage involves defining and designing a manufacturing process capable of consistently producing CBG isolate of the required quality. The goal of process design is to establish a process that will perform reliably in routine commercial production. Key activities include selecting appropriate production equipment and facilities, and identifying critical process parameters (CPPs) and critical quality attributes (CQAs) that affect CBG quality. Sound scientific experimentation (often at lab or pilot scale) is used to build understanding of the process; for example, Design of Experiments (DoE) studies and risk assessments (per ICH Q9 principles) help pinpoint which input variables (e.g. raw material properties or process settings) significantly impact output quality. Any risks identified (such as sensitivity of CBG to temperature or the potential for cross-contamination) are mitigated through process controls and safeguards. By the end of process design, a suitable process flow is defined along with defined control strategies to ensure that the CBG API will meet all specifications.
-
Installation Qualification (IQ): Before running the process, all manufacturing equipment and utility systems must be installed and configured correctly according to design specifications and manufacturer’s recommendations. Installation Qualification is the documented verification that the equipment as installed (or any modifications) complies with the approved design and user requirements. In practice, this means confirming that each vessel, chromatographic system, reactor, etc., is assembled with the proper materials, is calibrated, and is hooked up as intended (e.g. correct piping and instrument installation). According to FDA process validation guidance (aligned with ICH Q7), proper facility and equipment design and commissioning are prerequisites for process qualification. Successful IQ ensures the physical setup is suitable for subsequent operational testing and production use.
-
Operational Qualification (OQ): OQ is the next qualification step where equipment and systems are tested to verify they operate as intended across all anticipated operating ranges. In other words, OQ provides documented evidence that the installed equipment performs within the predetermined limits and tolerances under no-load and simulated load conditions. This involves challenging the equipment controls, settings, and alarms – for example, running a centrifuge through its full speed range, testing temperature probes across their range, and checking control system interlocks. The operational qualification confirms that utilities and equipment meet the operational requirements of the CBG process (e.g. a solvent purifier yields the required purity, a reactor achieves target temperatures) before actual product is made. By completing OQ, the manufacturer ensures that the process equipment will function reliably and consistently under the defined process parameters.
-
Performance Qualification (PQ): Once IQ and OQ are completed, Performance Qualification (sometimes called Process Performance Qualification, PPQ, in FDA terminology) is conducted. PQ is the documented verification that the equipment, utilities, and entire process as integrated can perform effectively and reproducibly to produce the CBG isolate meeting its quality specs on a consistent basis. Here, the process is executed under actual production conditions (using qualified personnel, raw materials, and at nominal batch sizes) to demonstrate that all CQAs are consistently achieved. Typically, multiple consecutive batches of CBG isolate are manufactured and tested. Critical process parameters are closely monitored to confirm the process remains in control. All resulting batches are analyzed to ensure they meet predefined specifications (purity, potency, impurity levels, etc.). Only if the process yields acceptable product consistently (with any deviations investigated and resolved) will it be considered validated for routine production. Successful completion of PQ, with an approved protocol and report, indicates the process can be scaled for commercial manufacturing with confidence in its robustness.
-
Cleaning Validation: In multi-use facilities or whenever equipment is used for different compounds, cleaning validation is essential to prevent cross-contamination. Cleaning procedures for the CBG process equipment must be proven effective in removing residues of CBG or any other products to acceptable levels. ICH Q7 emphasizes that cleaning validation should be directed at worst-case scenarios – equipment or steps where carryover poses the highest risk to API quality. A representative worst-case chemical (often the hardest to clean or most potent compound made in the equipment) is chosen for the validation. For cannabinoids, one major concern is carryover of psychoactive or potent impurities (for instance, trace Δ9-THC carryover into a CBG batch would be highly undesirable). Thus, robust cleaning validation is critical. The validation involves developing sampling methods (swabbing surfaces, rinse samples, etc.) and analytical tests sensitive enough to detect trace residues. The cleaning process is then executed and samples from equipment surfaces are tested to ensure any remaining residues are below pre-established limits based on potency/toxicity. Both the FDA and ICH Q7 guidance expect that non-dedicated equipment can be used only if validated cleaning prevents cross-contamination. Effective cleaning validation for CBG isolate manufacturing means each batch is not at risk of contamination from previous products or batches.
-
Continuing Process Verification: Process validation does not end after the initial qualification batches – it continues throughout the commercial production life of the CBG API. ICH Q7 (in alignment with ICH Q10 and modern PV practices) expects an ongoing program of monitoring critical process data and product quality, often referred to as Continued Process Verification (CPV) or ongoing process verification. The FDA’s process validation guidelines describe Stage 3 as the period of continued verification, where manufacturers gain ongoing assurance during routine production that the process remains in a state of control. In practice, this means establishing a monitoring plan: collecting data on critical parameters (e.g. reaction temperatures, pressures, potency results, impurity levels) for each batch, and using statistical tools to detect any drift or trends. Any signals or deviations trigger investigations and corrective actions to maintain control. Over time, the accumulated data also feed into Product Quality Reviews (annual reviews) that evaluate whether the process is still performing as validated. This continual verification is especially important for a novel API like CBG, where initial data may be limited – ongoing monitoring will quickly highlight if any aspect of the process needs adjustment. The outcome is a high level of assurance that each batch of CBG isolate continues to meet quality requirements consistently over the product’s lifecycle.
-
Revalidation: Changes are inevitable over the life of a process – for example, equipment upgrades, process optimization, new raw material sources, or scale-up to larger batch sizes. Whenever significant changes are made that could impact the process or product quality, an evaluation must be done to determine if revalidation is required. Regulatory guidelines (FDA, EMA, and ICH Q7 Q&A) state that changes to critical processes, parameters, or quality attributes demand re-assessment of the validation status. For instance, if a new filtration system is introduced or a critical drying parameter is altered for CBG isolate production, the manufacturer should perform impact assessments (using change control procedures) and likely run additional qualification or process confirmation batches. The scope of revalidation can range from repeating specific qualification tests to a full PQ repeat, depending on the change. The rationale and any supporting data (e.g. smaller-scale studies showing no impact) should be documented. By requiring revalidation after significant changes, ICH Q7 ensures that the high level of quality assurance is maintained and that any modifications do not adversely affect the CBG API’s purity, potency, or other critical qualities.
Connected Source Citations: The above process validation steps and concepts are grounded in ICH Q7 GMP guidance for APIs, FDA Process Validation guidance, and industry best practices. These sources emphasize that process validation is a lifecycle endeavor – starting from process design and equipment qualification through to continual monitoring – all aimed at ensuring a consistent, high-quality API output.
Stability Testing for CBG Isolate (ICH Q1 Guidelines)
Stability testing is the program that evaluates how the quality of the CBG isolate API varies with time under the influence of environmental factors like temperature, humidity, and light. The data from stability studies are used to establish a retest period or shelf life for the API and to recommend storage conditions. Because CBG is a novel cannabinoid with limited historical stability data, a comprehensive stability protocol is crucial. ICH has a series of guidelines (Q1A–Q1F) that outline the requirements for stability testing of new drug substances. In accordance with these, the following types of stability studies should be conducted for CBG isolate:
-
Long-Term (Real-Time) Stability Testing: Real-time stability studies involve storing CBG isolate under its expected label storage conditions (for example, a typical condition for many APIs is 25 ± 2 °C and 60 ± 5% RH for long-term, unless CBG requires cold storage) and testing the material at set intervals over an extended period. ICH Q1A(R2) requires that long-term (also called real-time) testing cover a minimum of 12 months on at least three primary batches, and continue for the full proposed retest period of the API. For instance, a CBG batch might be tested at 0, 3, 6, 9, 12, 18, 24 months, etc., for critical stability-indicating parameters (assay purity, degradation products, appearance, etc.). These real-time data demonstrate the actual shelf-life of CBG under normal storage. They are essential for confirming any provisional shelf-life that was estimated from accelerated studies. Real-time testing also reveals any long-term degradation products that may form and ensures the CBG isolate remains within specification throughout its shelf life.
-
Accelerated Stability Testing: Accelerated stability studies are carried out at exaggerated storage conditions (e.g. 40 ± 2 °C and 75 ± 5% RH for 6 months, per ICH Q1A standard conditions) to speed up the chemical and physical degradation processes. The idea is to simulate and predict the API’s stability over a longer period in a shorter time. Data from accelerated testing, combined with real-time data, can be used to extrapolate the CBG’s shelf life or establish interim retest periods. For example, if CBG shows minimal degradation after 6 months at 40 °C/75% RH, one could be confident in a shelf life of at least 24 months at ambient conditions, provided real-time data support it. However, if significant changes (e.g. notable assay loss or high degradation product formation) occur under accelerated conditions (ICH defines “significant change” thresholds for APIs), it may indicate that CBG is heat or humidity sensitive and thus might require a shorter shelf life or special storage (such as refrigeration). Accelerated studies also help identify likely degradation pathways; for instance, they may reveal if CBG tends to hydrolyze, oxidize, or otherwise break down under stress. It should be noted that while accelerated studies are predictive, not all degradation observed at stress conditions will occur under real conditions (e.g. physical changes like polymorph conversion might not extrapolate). Therefore, accelerated testing is a useful tool for rapid insight, but it must be supported by real-time data.
-
Stress Testing (Forced Degradation): In addition to formal stability storage conditions, stress testing (also called forced degradation studies) should be performed on CBG isolate to understand its intrinsic stability. ICH Q1A(R2) recommends stress testing of the drug substance to identify likely degradation products and establish degradation pathways. This typically involves exposing CBG to more extreme conditions than accelerated tests, such as high heat (e.g. 50–60 °C), high humidity (e.g. ≥75% RH), intense light, oxidizing conditions (e.g. hydrogen peroxide), and a range of pH (acidic and basic solutions). For each stress condition, samples of CBG are analyzed to see how much and what type of degradation occurs. For example, in strong acid or base, does CBG degrade rapidly? Under oxidative stress, does it form any specific oxidized impurity? These studies are typically done on a small scale and are not meant to determine shelf life, but rather to challenge the molecule’s stability to glean its chemical behavior. The information from stress testing is invaluable for developing and validating stability-indicating analytical methods – methods capable of distinguishing and quantifying degradation products. It also helps in understanding what degradation products might need to be monitored in long-term studies and if any special protective measures (like antioxidants in the formulation or inert gas purging in containers) are needed. In summary, stress testing of CBG provides a worst-case insight into how CBG might break down, ensuring no surprises during real storage and that all potential impurities are accounted for in the specifications.
-
Photostability Testing: Photostability is a specific aspect of stability focused on the effects of light exposure. Cannabinoids like CBG are often light-sensitive (many develop color or potency loss with light), so ICH Q1B guidelines mandate evaluating photostability. Photostability testing can be considered a subset of stress testing, but it is typically done as a dedicated study following Q1B conditions. For confirmatory photostability studies, CBG samples should be exposed to a defined amount of light – ICH Q1B specifies an exposure of not less than 1.2 million lux hours of visible light and 200 watt-hours/square meter of UVA light. This intense light exposure (essentially equivalent to prolonged sunlight/room light) is designed to induce any photo-degradation that might occur. During photostability testing, one set of CBG samples is exposed to light, while another set is protected (wrapped in foil) as a dark control. After exposure, the samples are examined for changes in appearance, assay, and degradation products. If CBG is photolabile, the exposed sample may show new impurities or a drop in assay compared to the dark control. The results will determine if CBG needs protection from light (for example, requiring storage in light-resistant containers). Many drug substances do require such labeling (“protect from light”) if they are unstable when exposed to light. Given that CBD and Δ9-THC are known to undergo photodegradation to some extent, similar behavior is evaluated for CBG. The photostability study ensures that appropriate precautions (amber glass bottles, opaque packaging) can be implemented for CBG isolate if necessary, and it confirms that light exposure during manufacturing or storage will not compromise product quality.
Connected Source Citations: The stability protocols above are based on ICH Q1 guidelines for stability testing of new drug substances and reflect the practices needed to justify an API’s expiration or retest dating. Implementing a full battery of long-term, accelerated, stress, and photostability tests for CBG isolate will yield the data required to assign a suitable retest period and storage conditions. Notably, regulatory expectations are that an API’s expiry/retest date be supported by real-time stability data, with accelerated studies aiding in the initial predictions.
Lessons from CBD and CBG-Specific Considerations
Experience gained from the development and validation of CBD (cannabidiol) as an API provides a valuable starting point for CBG, given the structural similarities between these cannabinoids. Both CBG and CBD are non-psychoactive cannabinoids with related chemical frameworks, so it is reasonable to anticipate some overlap in their manufacturing and stability challenges. For instance, the importance of robust cleaning validation was underscored in CBD production, where any cross-contamination (especially with psychoactive THC or other cannabinoids in multi-use facilities) had to be strictly prevented. The same principle carries over to CBG: equipment must be either dedicated or proven clean such that no residual carryover of other substances occurs. Another lesson from CBD is the critical need for meticulous documentation and Standard Operating Procedures (SOPs) throughout the process. In CBD API manufacturing, detailed SOPs and batch records were developed to ensure consistency in every step – from extraction and decarboxylation to purification and packaging. Those GMP documentation practices apply equally to CBG. A strong quality system (per ICH Q7 and Q10) with change control, deviation management, and employee training will form the backbone for both CBD and CBG production. By leveraging the established procedures and quality systems from CBD, a manufacturer can avoid “reinventing the wheel” and more quickly achieve a compliant process for CBG. In summary, the general cGMP controls – cleaning, equipment qualification, process monitoring, documentation – that were proven in CBD operations are directly transferable to CBG API production.
Despite these similarities, CBG also presents unique challenges that differentiate it from CBD, necessitating special focus in the validation and testing program. One major difference lies in the relative lack of historical data: CBD has been widely studied (and even approved in medicines like Epidiolex®), whereas CBG’s profile is less understood because, until recently, CBG was typically present only in small quantities in cannabis plants. This paucity of prior knowledge means that for CBG, the development team cannot rely on extensive literature or pharmacopeial monographs for stability insights or processing parameters – instead, they must generate that data themselves. For example, stability testing for CBD had revealed that CBD is susceptible to degradation when exposed to certain conditions: it can oxidize to form hydroquinone derivatives, it can form a quinone upon exposure to air in basic conditions, and under acidic conditions CBD can cyclize into Δ9-THC and other cannabinoids. These findings guided formulators to avoid strong acids and prolonged light/heat for CBD products. In the case of CBG, one might expect analogous vulnerabilities (being an organic cannabinoid, CBG could also be prone to oxidation or light-induced changes), but it is imperative to confirm this experimentally. The degradation pathways of CBG must be elucidated via the stress studies described earlier. Perhaps CBG will show a different dominant impurity profile than CBD – for instance, while CBD can cyclize to THC, CBG might instead isomerize or break down into other minor cannabinoids or unknown compounds that need identification. Without prior precedent, the CBG stability program must be comprehensive, and methods must be validated to detect any potential degradation product. This highlights a point of divergence: whereas CBD’s stability profile is relatively mapped out in literature, CBG’s stability (effects of temperature, pH, oxidation, light, etc.) is largely uncharted, requiring extra diligence in analysis and possibly tighter control of environmental factors until confidence in its stability is built.
Another area of difference is in the process chemistry and yield. CBG is the precursor (“mother cannabinoid”) to CBD and THC in the biosynthetic pathway, and in harvested cannabis plants much of the CBG (as CBGA) has typically converted to other cannabinoids. Consequently, producing CBG isolate at commercial scale may involve different tactics: for example, using cannabis chemovars bred for high CBG content, or isolating CBGA and then decarboxylating, or even synthetic and biosynthetic production methods. These production differences mean the impurity profile of CBG could differ from CBD. CBD isolate processes often contend with removing traces of other cannabinoids and terpenes; with CBG, there might be higher relative levels of residual CBGA or related compounds to control. Each unique impurity needs its own analytical method and specification limits, which again must be validated as part of the quality program. The process validation for CBG has to accommodate these differences – for instance, critical process parameters might include the decarboxylation temperature to convert CBGA to CBG, which has no exact analog in a CBD isolate process (CBD comes from CBDA decarboxylation, but the kinetics and optimal conditions might differ). Such parameters would be studied during CBG process design to ensure efficiency and minimal byproduct formation.
In summary, while the framework and lessons from CBD give CBG a head start – illustrating the importance of GMP rigor in cleaning, consistent process execution, and thorough stability testing – CBG demands its own data-driven validation. The lack of extensive historical use means that the validation and stability program for CBG isolate must be especially robust and closely aligned with ICH guidelines, leaving no stone unturned. By rigorously applying GMP principles and learning from CBD’s stability and process idiosyncrasies, manufacturers can confidently establish that CBG isolate meets the same high standards of quality, purity, and safety expected of any API. The end result of this effort is a CBG API that is fully supported by scientific data – its process is validated to consistently produce quality product, and its stability profile is well-understood to ensure patients and regulators can trust its efficacy throughout its shelf life.
Connected Source Citations: The discussion above references the known chemical behavior of CBD and the historical scarcity of CBG data to highlight the rationale for CBG-specific precautions. By following ICH Q7 compliant validation practices and ICH Q1 stability guidelines, the unique challenges of CBG can be managed such that the cannabinoid’s promise as a pharmaceutical ingredient is realized with assured quality. All changes and lessons are documented within a robust quality system, ensuring that both regulatory expectations and scientific best practices are met in bringing this novel cannabinoid API to market.
The complete guide to the ICH Q7 guidelines
Cannabigerol (CBG): A Comprehensive Review of Its Molecular Mechanisms and Therapeutic Potential
Q7 Good Manufacturing Practice Guidance for Active Pharmaceutical Ingredients Guidance for Industry
Process Validation: General Principles and Practices
“How to do” Document
Q 1 A (R2) Stability Testing of new Drug Substances and Products
[PDF] Q1A(R2) Guideline – ICH
In the right light: What ICH photostability tests are all about
Stability, biofunctional, and antimicrobial characteristics of cannabidiol isolate for the design of topical formulations – Soft Matter (RSC Publishing) DOI:10.1039/D3SM01466E